Scientists at Goethe University Frankfurt, together with colleagues from Darmstadt, Heidelberg, Oxford and Dundee (UK), have investigated how the fit of potent inhibitors to their binding sites can be optimised so that they engage longer with their target proteins. Long target residency has been associated with more efficient pharmacological responses, for instance in cancer therapy. The result: High resolution structures revealed that when the interaction between the inhibitors and the target proteins lasts particularly long, the target proteins „nestle“ against the inhibitors. In future, the researchers want to use computer simulations to predict the residence time of inhibitors during drug development.
Many anti-cancer drugs block signals in cancer cells that help degenerated cells to multiply uncontrollably and detach from tissue. For example, blocking the signalling protein FAK, a so-called kinase, causes breast cancer cells to become less mobile and thus less likely to metastasise. The problem is that when FAK is blocked by an inhibitor, the closely related signalling protein PYK2 becomes much more active and thus takes over some of FAK’s tasks. The ideal would therefore be an inhibitor that inhibits both FAK and PYK2 in the same way for as long as possible.
An international team led by the pharmaceutical chemist Prof. Stefan Knapp from Goethe University has investigated a series of specially synthesised FAK inhibitors. All inhibitors bound to the FAK protein at about the same rate. However, they differed in the duration of binding: The most effective inhibitor remained bound to the FAK signalling protein the longest.
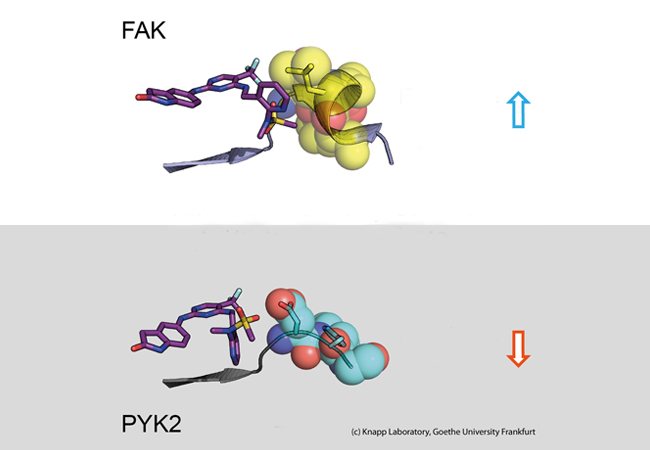
Using structural and molecular biological analyses as well as computer simulations, the research team discovered that binding of inhibitors that remain in the FAK binding pocket for a long time induce a structural change. Thus, through binding of these inhibitors, FAK changes its shape and forms a specific, water-repellent structure at contact sites with the inhibitor, comparable to an intimate embrace.
The closely related protein PYK2, on the other hand, remained comparatively rigid, and although the most effective FAK inhibitor also blocked PYK2, its effect was significantly weaker due to quickly dissociating inhibitors from the binding site. Interestingly, computer simulations were able to predict the kinetics of binding very well, providing a method for accurate simulation of drug dissociation rates for future optimisation of drug candidates.
Prof. Stefan Knapp explains: „Because we now have a better understanding of the molecular mechanisms of the interaction of potent inhibitors of these two kinases, we hope to be able to use computer simulations to better predict drug residence times of inhibitors and drugs candidates in the future. So far, little attention has been paid to the kinetic properties of drug binding. However, this property has now emerged as an important parameter for the development of more effective drugs that are designed to inhibit their target proteins – as in the case of FAK and PYK2 – not only potently but also for a long time.“
Publication: Benedict-Tilman Berger, Marta Amaral, Daria B. Kokh, Ariane Nunes-Alves, Djordje Musil, Timo Heinrich, Martin Schröder, Rebecca Neil, Jing Wang, Iva Navratilova, Joerg Bomke, Jonathan M. Elkins, Susanne Müller, Matthias Frech, Rebecca C. Wade, Stefan Knapp: Structure-kinetic relationship reveals the mechanism of selectivity of FAK inhibitors over PYK2. Cell Chemical Biology https://doi.org/10.1016/j.chembiol.2021.01.003
This work was carried out within the framework of the public-private partnership K4DD (Kinetics for Drug Discovery) of the Innovative Medicinces Initiatives (IMI). https://www.k4dd.eu/home/